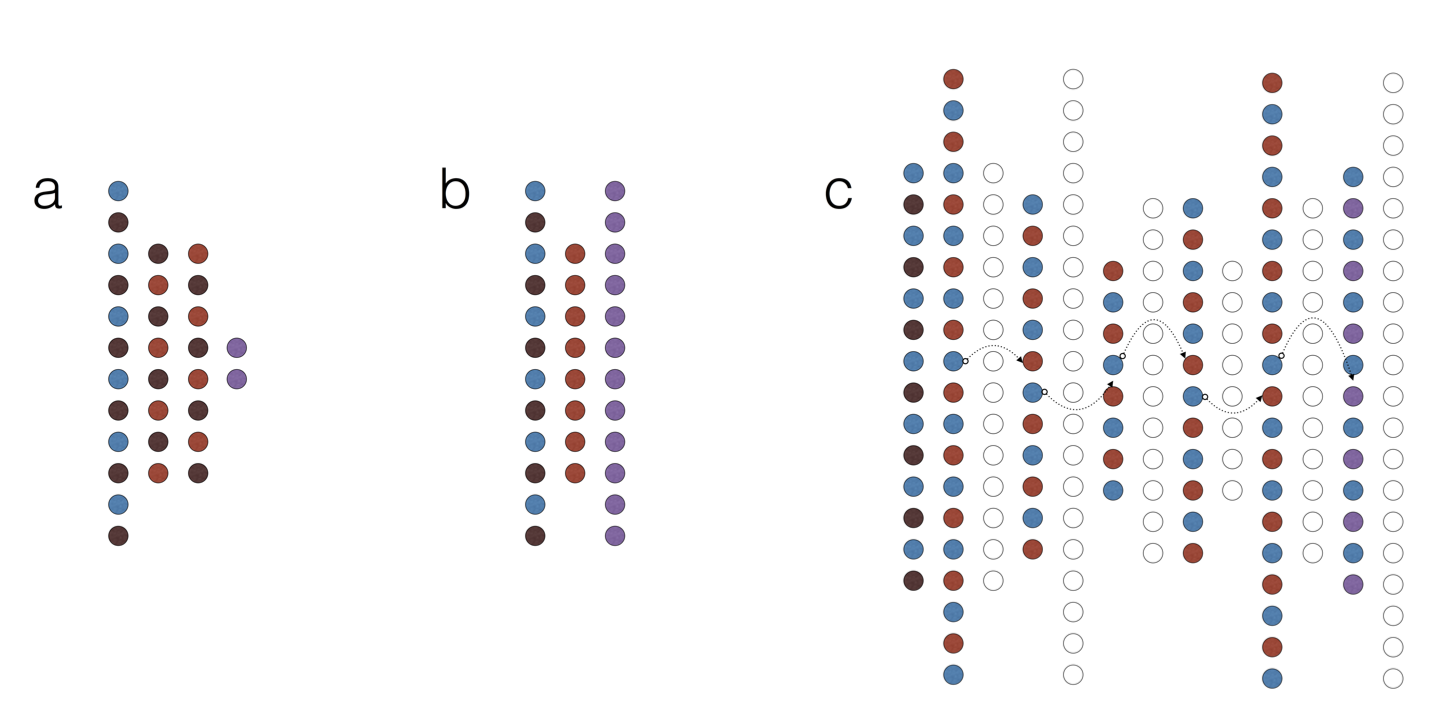Experimental projects |
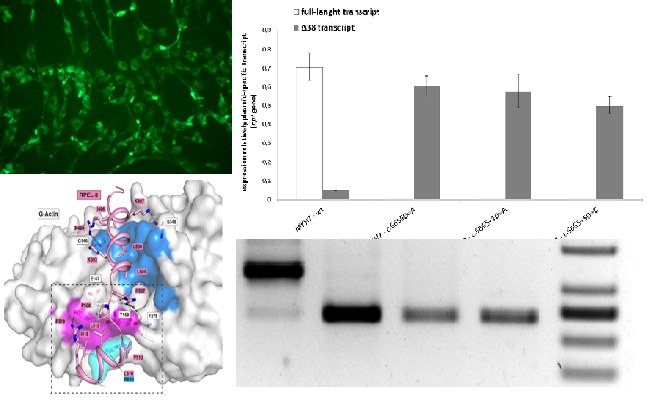
Functional analysis of nucleotide variants of unknown clinical significanceOne of the main direction of the Laboratory work is a functional analysis of nucleotide variants of unknown clinical significance which are found in high throughput sequencing of DNA of patients with rare inherited disorders. Interpretation of genetic variants consequences and its relation to the patient's phenotype is a challenge in a huge number of cases, and only functional analysis of such genetic alterations could answer this question. A vast majority of methods are used to confirm a pathogenic role of the variant: RNA analysis, immunofluorescence, reporter assays, enzymatic activity measurements. Everything depends on the affected protein properties as well as the way of damage by which a mutation disrupts a gene. We performed functional analysis either on in vitro systems or on patient's primary cultures of cells which subsequently are stored in the BioBank at the Research Centre for Medical Generics and could be used for additional analyses in case of novel symptoms will develop in patient or novel information of gene function will be available by scientific community which require novel assays. Among the cases which were resolved with the aid of functional analysis performed in the Laboratory, a discovery of novel genes which were not previously linked to human disease, KRT25, PHACTR1; functional analysis of deep intronic nucleotide variants in B4GALT7, MYH7, PALB2, and others; assessment of missense mutations impact on function of COL6A3, DHTKD1; and many more. This field of the Laboratory work provides a link between fundamental human biology and clinical genetics to broaden our understanding of etiopathological mechanisms of human disease as well as human genes function.
|
Long non-coding RNAs and antisense transcriptionNatural antisense transcripts (NATs) represent RNAs containing sequences that are complementary to other endogenous RNAs, including pre-mRNAs. They can be transcribed in cis from opposing DNA strands at the same genomic locus (cis-NATs), or in trans from separate loci (trans-NATs). Recently it has been shown NATs have been implicated in many aspects of eukaryotic gene expression including genomic imprinting, RNA interference, translational regulation, alternative splicing, X-inactivation and RNA editing. Moreover, there is growing evidence to suggest that antisense transcription might have a key role in a range of human diseases. Although>20% of human genes might form cis-NATs pairs, the extent to which they are involved in antisense regulation is unknown. Special interest is represented with a hypothesis about relation between the level of antisense transcription throughout the genome and the progression of malignant tumors. To study this relationship we are investigating sense-antisense pairs that significantly differ in their expression levels in normal and tumor cells in human. This provides the basis for understanding of one of the major ways of regulation of a transcription - antisense RNA-dependent gene expression regulation in normal and tumor cells. Now we are studying the specific examples of different NATs in human genome. We extend our research from the study of impact of the NATs in tumorigenesis to the investigation of the role of other types of non-coding RNAs in gene expression regulation in human. Here we focus on the interaction of different types of RNA which could have an impact on global gene expression regulation. |
A comparative study of structure and function of KCTD proteinsRecently in our laboratory the novel potential tumor suppressor gene KCNRG had been discovered in the study of the B-cell chronic lymphocytic leukemia cause. The protein appeared to contain a single conserved domain, T1 potassium channel tetramerization domain, which is required for the interactions of voltage-gated potassium channels protein subunit. KCNRG has been shown to negatively regulate potassium currents in vitro as well as suppress the proliferation and activate the apoptosis in cancer cell lines. Subsequent in silico whole-genome search revealed the whole family of only T1 domain-containing proteins in human genome called KCTD family. The main goal of our study was to functionally characterize the family of KCTD proteins. We are focused on the interactome of KCTD family and their diverse potential functions. |
Development of new diagnostic methods for medical geneticsOur group also help clinical geneticists to diagnose different rear hereditary disorders by development of novel molecular diagnostics. The list of genetic disorders includes: Clouston's hidrotic ectodermal dysplasia, facioscapulohumeral muscular dystrophy Landouzy-Dejerine, cistic fibrosis, hereditary hypotrichosis, etc. |
Bionformatics projects |
Applying Deep Learning To Integrate Deleteriousness Prediction Scores For Whole-Exome SNV StudiesNowadays computational biology is becoming increasingly data-driven. On the one hand, this opens new possibilities for statistical modelling of complicated biological systems. On the other, extracting meaningful abstractions from a data flood is no small feat. In our laboratory we work on the very frontier of statistical inference in various fields of biomedical science. We research, develop and apply new deep-learning and Bayesian methods to analyse amino acid substitutions, text mining and biological networks to achieve great generalisability. Our models, such as DeepEVS (Korvigo et al., 2017, pre-print: https://doi.org/10.1101/126532) demonstrate state-of-the-art performance at predicting causative mutations in proteins.
Fig. Network types. Schematic representation of basic deep learning models used in this study. (a) A multilayer perceptron (MLP). (b) A shallow denoising autoencoder (dAE). (c) Connecting dAEs into a stacked denoising autoencoder (sdAE); notice that each individual dAE learns to reconstruct the latent representation from the previous one (data stream is represented by arrows). Colours encode layer functions (combinations are possible): blue - input, light-red - latent, dark-red - dropout (noise), purple - output, hollow - discarded. |
Analysis of human miRNA - mRNA interactomemiRNAs are a family of small RNA molecules that regulate a wide range of biological processes through post-transcriptional regulation of gene expression. Despite years of research, the principles of miRNA targeting are incompletely understood. The recent high-throughput techniques CLASH (Helwak et al., 2013) and CLEAR-CLIP (Moore et al., 2015) allow to revealed miRNAs ligated to their endogenous mRNA targets in HEK293 cell line and in human hepatoma cells respectively. We apply bioinformatics methods for proceeding and understanding insights into miRNA – mRNA interactions in the cell. To accomplish this, we observed the associations of the expression level of miRNAs and mRNAs. Our aim is to understand the role of miRNA and identification of their targets that alter gene expression and could influence many aspects of health and human disease. |
Analysis of RNA modificationThere are more than 100 different RNA modifications. Recent technical advances have revealed modification of messenger RNAs with N6-methyladenosine (m6A), 5-methylcytosine (m5C) and pseudouridine (Ψ). RNA modification play an role in RNA stability, translation efficiency and genetic recoding (Gilbert, 2016). There are available whole-transcriptomic data about several types of RNA modifications, but frequency of pathogenic mutations in the sites of modifications is not studied yet. The goal of our work is an analysis of possible pathogenecity of mutations in the sites of RNA modification. |
Finished projects |
Natural history of kidney at the pre-transplantation period and ways to improve the quality of donor organsGlobal organ shortage is the crucial point of transplantation nowadays. Usage of expanded criteria donors represents reliable source of donor organs, making transplantation more accessible for patients with end stage organs failure. Ischemia-reperfusion injury followed by the activation of programmed cell death scenarios remains the main obstacle in utilization of marginal grafts. Programmed cell death often leads to life threatening complications in posttransplant period. The main goal of the study is to investigate damading processes mechanisms, ways of preservation and improvement of viability of donor kidney. We also work on the ntisense gene therapy which could provide a therapeutic tool, capable to improve quality of grafts and, consequently, transplantation outcomes. This project is conducted in collaboration with St. Petersburg State Medical University named after Academician I.P. Pavlov. |
Microarray analysisMicroarray gene expression profiling offers an opportunity for genome-scale, quantitative evaluation of gene expression studies by simultaneously measuring expression levels for thousands of genes. Nevertheless the discrepancies in probe sets data remained to be wide-spread phenomenon which is usually underestimated or dissembled. So the correct annotation of probe sets remains the actual problem. The man goal of the study is to determine ways of application which could help correctly represent the real gene expression profile in human transcriptome. We also investigate the approaches to study cis- and trans-antisense interactions in human transcriptome using expression microarrays. |
Bioinformatic analysis of regulatory antisense RNA-RNA interactionsWe are developing a tool and a database for prediction of antisense interactions between transcripts. The number of possible short antisense interactions between transcripts is large. For alignment lengths less than 10 any transcript has an antisense interactions with almost any other RNA. However in reality only few of these interactions result in regulation of gene expression. So, the main challenge in this project is to identify the most promising antisense hits for a given query. Currently we utilize two main approaches�expression and conservation analyses. You could find the online tool here: http://assa.generesearch.ru |
RANDTRAN: random transcriptome sequence generatorRandom transcriptome generation is a frequently used method in modern biopenrmatics researches. However algorithms of its generation are not widely discussed in the literature and the concept for the design of random transcriptome is often limited to fixed mononucleotide composition frequency. Our aim was to make the program for random transcriptome generation that takes into account the structure of transcript (5'-UTR, CDS, 3'-UTR, or ncRNA) with its own dinucleotide and trinucleotide (codon) intrinsic frequencies for different species. The program �RANDTRAN� has user-friendly interface and flexible options to create your own random transcriptome. It used a standalone .txt frequency file (2KB) that defined a lot of parameters for specific organism. For the moment the program is packaged with 23 files (for 11 eukaryotes and 12 bacteria and archea species). The frequency file contains a lot of parameters described special features, such as dinucleotide and trinucleotide frequencies for each transcript region, length distribution data of each part of transcript, quantity of coding and non-coding transcripts. The advantage of our program is to allow changing of above arguments or creating new frequency file that gives a user the opportunity to generate the transcriptome with necessary characteristics. The detailed manual is attached. |